


































亀田総合病院腎臓高血圧内科部長
鈴木 智

2006年聖マリアンナ医科大学卒業。2016年聖マリアンナ医科大学大学院修了。千葉東病院アレルギー科,聖マリアンナ医科大学腎臓高血圧内科,筑t波大学腎血管病理学特別研究生を経て,2018年より現職。2022年から福島県立医科大学大学院研究科臨床疫学分野の博士研究員を併任。腎炎,腎病理をサブスペシャリティとし,ANCA関連血管炎,MGRSなどに力を入れている。
◉ANCA関連血管炎を疑うことは難しくない。
◉ANCA関連血管炎の腎病理像は多彩である。
◉ANCA関連血管炎の治療は急速に進歩しており,ステロイドを少なくすることや,早期に中止にできる可能性がある。

ANCA関連血管炎(antineutrophil cytoplasmic antibody-associated vasculitis:AAV)は,Chapel Hill Consensus Conference(CHCC)で提唱されたChapel Hill分類で定義される,主に小型から中型の血管を障害する自己免疫性炎症性疾患であり,抗好中球細胞質抗体(ANCA)の出現を特徴とする疾患群である。AAVは顕微鏡的多発血管炎(microscopic polyangiitis:MPA),多発血管炎性肉芽腫症(granulomatosis with polyangiitis:GPA),および好酸球性多発血管炎性肉芽腫症(eosinophilic granulomatosis with polyangiitis:EGPA)の3疾患に分類される。そのほかには,MPAの中で腎病変のみを認める腎限局型血管炎(renal limited vasculitis:RLV),薬剤性血管炎も含まれる1)。これらの疾患は進行性で,多臓器に障害を引き起こす。特に腎臓や肺の障害は患者の生命予後に直結するため,迅速な診断と治療介入が不可欠である。
本疾患群は稀少疾患であり,日本における推定有病率はおよそ6000〜7000人に1人とされている。これまでは,初期症状が非特異的であることも少なくないため,早期診断が困難であった。しかし,近年は,疾患啓発が進んだことや,重要な検査である血清のANCAの測定が簡便となり,施設によっては施設内で検査をし,その日のうちに結果を確認することができるため,以前より早期診断ができるようになってきた。
加えて,ステロイド中心の治療から,免疫抑制薬や補体に対する治療が登場したことにより,近年は寛解可能な疾患へと位置づけが変わってきた。そのため,短期的な生命予後や臓器予後の改善だけでなく,ステロイドや免疫抑制療法に伴う感染症を避けることや,生活習慣病管理を含めたADLやQOLの向上などの長期的な課題も重要となってきた。本稿では,AAVの基本的な病態,臨床症状から最新の診断・治療戦略について解説し,実臨床における診療の質向上の一助になることを期待している。
AAVは,稀少疾患に分類されるが,免疫関連疾患の中では頻度が比較的高い血管炎症候群である。日本におけるAAVの推定有病率は約15.2人/10万人,年間発症率は2.4〜3.0人/10万人程度とされている。欧州と比較すると,MPAの割合が高く,PR3-ANCAよりもMPO-ANCA陽性例が優位である点が特徴的であり,人種差や環境要因の影響が考えられている2)。わが国に多いMPAの平均発症年齢は71.1歳であり,AAVは腎障害の合併が多く,特に高齢者に初発した血尿ではANCA関連腎炎を念頭に置くことは重要である3)。加えて,腎炎を示す尿所見を伴い,数週から数カ月の経過で急速に腎不全が進行する「急速進行性腎炎症候群(rapidly progressive glomerulonephritis:RPGN)」 は生命予後に大きく関わるため,AAVの重要な合併症である。日本からの報告では,RPGNの原因として,AAVが67.9%と最も多く4),また,ANCAと抗糸球体基底膜抗体(抗GBM抗体)がともに陽性となる例が5.8%ある5)。抗GBM抗体型腎炎は,ANCA関連腎炎より重症度が高く,RPGNの場合は,MPO-ANCA,PR3-ANCAに加えて抗糸球体基底膜抗体の測定も重要となる。
AAVは,ANCAの出現を特徴とする自己免疫性小型血管炎である。遺伝的素因(HLAなど)があるところに,環境要因(感染症や薬剤など)が加わり,好中球が活性化する。ANCAは,活性化した好中球の表面に一時的に発現したミエロペルオキシダーゼ(myeloperoxidase:MPO)やプロテイナーゼ3(proteinase 3:PR3)に結合し,好中球の脱顆粒は活性酸素,プロテアーゼ,好中球細胞外トラップ(neutrophil extracellular traps: NETs)の放出につながり,内皮細胞を障害する。ケモカインとPR3とMPOの組織沈着は,自己反応性T細胞と単球の動員をもたらし,組織傷害を増大させ,血管壁の炎症・壊死性変化をきたす1)。この過程には,補体C5aの関与がきわめて重要であることが近年明らかとなり,C5a受容体阻害薬アバコパンの臨床応用につながっている。
AAVは,主にMPA,GPA,EGPAにわかれる。これらを分類するために,古くからWattsのアルゴリズムが使用されてきたが,3つの疾患の新しい分類基準が,2022年に米国リウマチ学会/欧州リウマチ学会(ACR / EULAR)から発表された(表1a 6),b7),c8))。この3つの分類基準を見ると,それぞれの疾患の特徴がわかる。MPAは,壊死性半月体形成性糸球体腎炎が高頻度にみられ,肺胞出血,間質性肺炎などの肺疾患の合併も多く,MPO-ANCA陽性が重視されている。GPAは,壊死性肉芽腫性炎症と血管炎を併存し,耳鼻咽喉科領域の症状(副鼻腔炎,中耳炎など)が初発症状となり,典型的には鼻→肺→腎の順番で進行することが多く,PR3-ANCA陽性が重視されている。EGPAは,好酸球増多症と喘息の病歴を有する患者に発症することが特徴であり,末梢神経障害(多発性単神経炎)や心筋炎など多彩な臓器病変を伴い,ANCA陰性例が比較的多く(約50%),ANCA陰性では腎炎の合併は稀である。

日本のコホートでも,この分類基準が検証され,Wattsのアルゴリズムに比べて,GPAとの分類不能が減り,MPAが増えた。最終的には477人の患者のうち,それぞれMPA(75.6%),GPA(9.9%),EGPA(10.7%),分類不能(6.1%)となり,MPAが圧倒的に多いことが再確認された9)。ただし,本分類基準は,小・中型血管炎と診断された患者を分類するために適用すべきであるため,血管炎類似疾患の除外をすることが重要となる。血管炎の鑑別疾患は後述する。
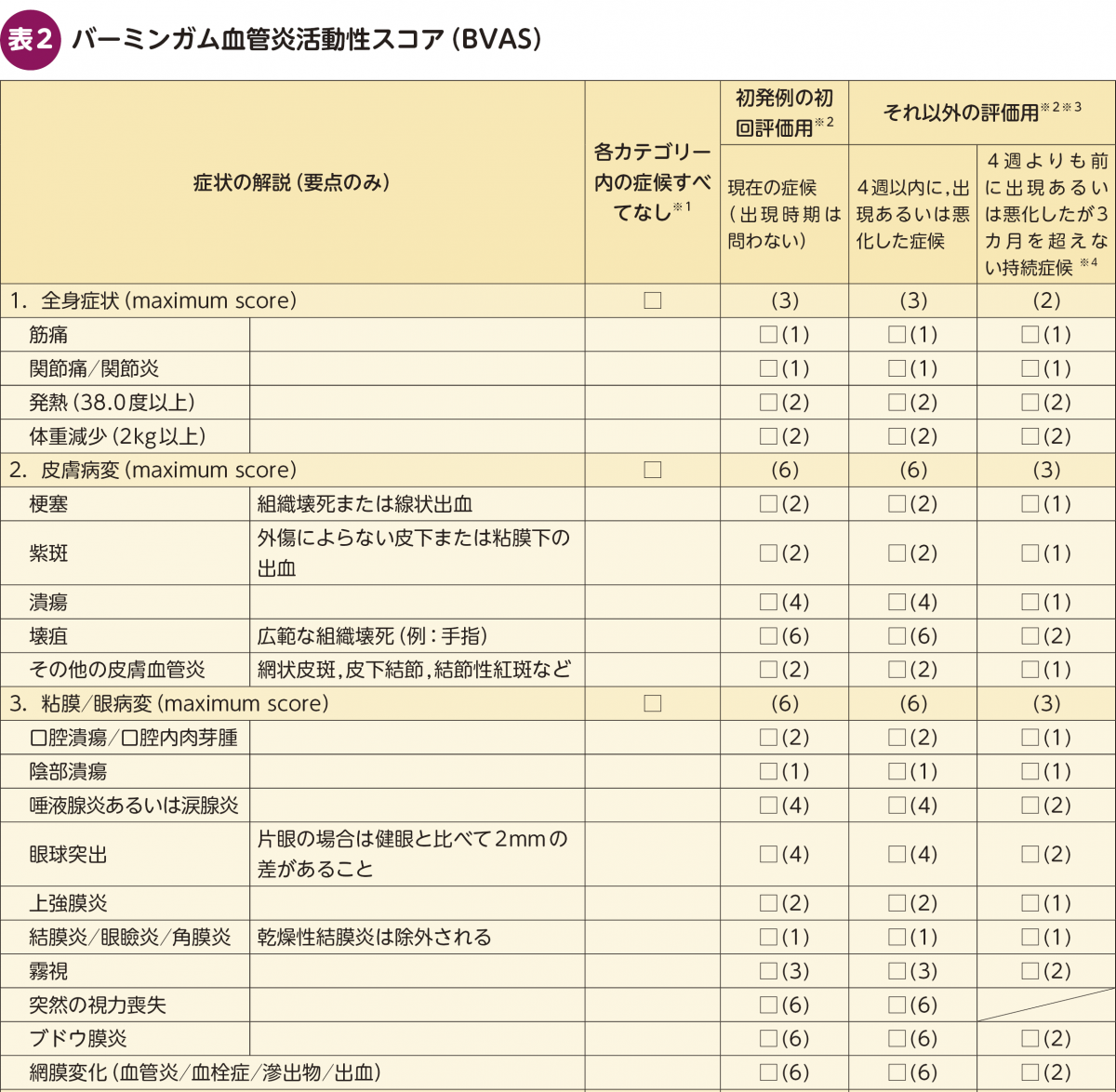
AAVは,様々な臓器に病変を起こしうるため,活動性の指標として,全身を評価するバーミンガム血管炎活動性スコア(Birmingham Vasculitis Activity Score:BVAS)がある。BVASは世界的に検証された血管炎の疾患活動性の指標である。血管炎の徴候および症状について,全身症状を含む臓器系(皮膚,粘膜/眼,耳鼻咽喉,胸部,心血管,腹部,腎,神経)の9項目で評価する。現在,BVAS ver3が使われることが多く,指定難病の申請にも使用されており,血管炎の全体像を理解する上でも内容を理解することは重要である(表2)10)。


プレミアム会員向けコンテンツです
→ログインした状態で続きを読む










